超高效液相色谱-串联质谱法测定茶叶中百草枯
百草枯是一种速效接触型灭生性除草剂,属于季铵盐类除草剂,使用遍及全世界130多个国家和地区[1]。百草枯极易溶于水,微溶于低相对分子质量的醇类,不溶于烃类溶剂,在酸性、中性溶液中稳定,在碱性溶液中易分解。随着百草枯的日益推广使用,其对人体健康和生态环境存在的严重危害在一定程度上已成为社会问题。百草枯对人、畜均有较强的毒性[2],目前尚无特效治疗方法。百草枯的高毒性和广泛应用性,使得建立快速、高选择性、高灵敏度的百草枯检测方法非常必要。检测的方法主要有气相色谱法或气相色谱-质谱法[3-4]、高效液相色谱法或高效液相色谱-串联质谱法[5-11]。
百草枯为强极性化合物,采用水浸法提取效率较高,一般对水提取液净化后直接进样分析,这样无法满足低含量残留的需求。若采用高温煮沸法进行浓缩,方法回收率低,前处理操作费时。本工作采用强酸性阳离子交换树脂进行吸附,氯化铵溶液进行洗脱,再在碱性条件下进行衍生反应,反应产物在C18色谱柱上分离后,通过串联质谱进行检测,能够获得准确、快速和高灵敏度的结果,并满足各项质控要求。
1 试验部分
1.1 仪器与试剂
WatersXevo TQ-S型超高效液相色谱-串联质谱仪;SUPELCO VISIPREP 24TMDL型固相萃取装置;Thermo Multifuge X3R型高速离心机;EYELASB-1100型旋转蒸发仪;NM100-SSCX强酸性阳离子交换树脂(0.15~0.25mm)。
百草枯标准储备溶液:1.00g·L-1,称取百草枯标准品(纯度不小于99.0%)0.010g,用甲醇溶解并定容至10mL,于-20℃冷冻保存。
百草枯标准溶液:2.0mg·L-1,移取1.00g·L-1百草枯标准储备溶液20μL至10mL容量瓶中,用水定容至10mL,于4℃冷藏保存。
甲醇、乙腈均为色谱纯,甲酸的纯度不小于95%,其他试剂均为分析纯,试验用水为超纯水(电阻率为18.25MΩ·cm)。
1.2 仪器工作条件
1)色谱条件Waters BEH UPLC C18 色谱柱(2.1mm×50mm,1.7μm),柱温40 ℃;流量为0.4mL·min-1;进样量3μL。流动相:A 为乙腈,B为乙腈(5+95)溶液(含体积分数为0.1%的甲酸和1 mmol·L-1 乙酸铵)。梯度洗脱程序:0~1.0min时,A 为0;1.0~1.5min时,A 由0升至90%,保持1.3min;2.8~2.9min时,A由90%降至0,保持1.1min。
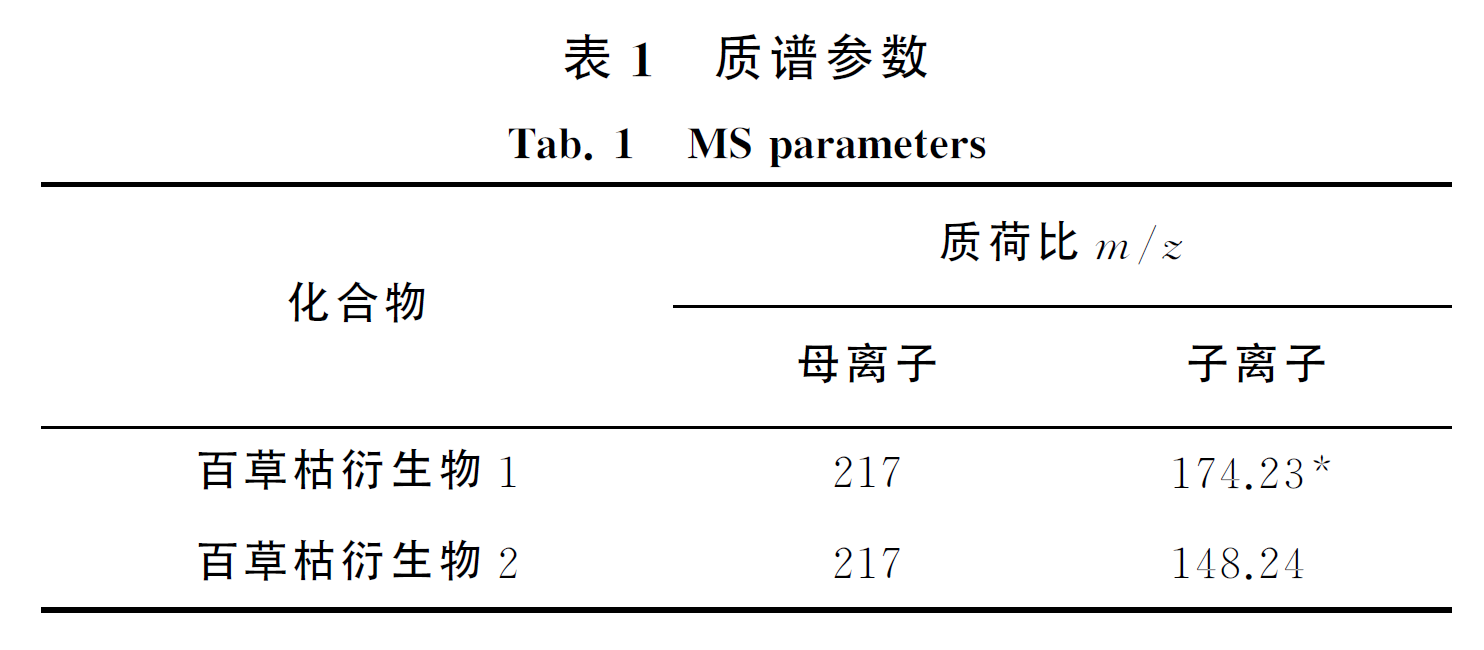
2)质谱条件 电喷雾离子源(ESI),正离子电离模式;毛细管电压3.0kV;离子源温度150℃;锥孔反吹气流量60L·h-1;脱溶剂气温度500℃,脱溶剂气流量1000L·h-1;碰撞池压力0.3Pa;锥孔电压66V,碰撞电压26V;扫描方式为多反应监测(MRM)模式,其余质谱参数见表1,其中“*”为定量离子。
1.3 试验方法
1.3.1 样品提取
称取均质茶叶样品(2±0.02)g至50mL一次性离心管中。加水13mL,使茶叶充分润湿,加入硫酸7mL,涡旋混匀,于80℃水浴中加热12h以上。取出,冷却至室温后,用水定容至50mL,以转速3500r·min-1离心5min,待用。
1.3.2 样品净化
NM100-SSCX强酸性阳离子交换树脂使用前需依次用3倍体积的甲醇和3倍体积的水活化,活化后的固相萃取(SPE)柱填料加2倍体积的水置于2~8℃冰柜中避光保存。
将普通定性滤纸剪成与2.5mL注射器内径大小相同的垫片置于注射器底部,加少量水抽干,使其与注射器底部紧密贴合,移取活化好的NM100-SSCX强酸性阳离子交换树脂填料填入注射器中,每个柱管内填充0.5mL待用(填料要均匀填充,防止气泡产生,上面要保留少量水,不要流干)。
移取样品提取液5mL过SPE柱(注意控制流量),用水淋洗至淋洗液呈中性,加5mol·L-1氯化铵溶液15mL洗脱,收集洗脱液于50mL离心管中。
1.3.3 衍生
向离心管中加入12mol·L-1 氢氧化钠溶液12mL,10g·L-1铁氰化钾溶液1mL,摇匀,待离心管中溶液冷却至室温后,加入二氯甲烷20mL,剧烈振摇0.5min,以转速3500r·min-1离心1min,移取下层溶液至150mL旋蒸瓶中,再加入二氯甲烷20mL重复提取一次,合并二氯甲烷层,于40℃水浴中减压旋蒸至干。用乙腈(1+9)溶液2mL超声溶解残渣后,过0.22μm有机系滤膜,按仪器工作条件进行测定。
1.3.4 0.1mg·L-1百草枯衍生物标准溶液的制备移取2.0mg·L-1百草枯标准溶液0.5mL于离心管中,加入5mol·L-1氯化铵溶液15mL,待离心管中溶液冷却至室温,加入二氯甲烷20mL剧烈振摇0.5min,以转速3500r·min-1离心1min,移取下层溶液至150mL旋蒸瓶中,再加入二氯甲烷20mL重复提取一次,合并二氯甲烷层,于40℃水浴中减压旋蒸至干。用乙腈(1+9)溶液10mL超声溶解残渣后,过0.22μm有机系滤膜,转入棕色储液瓶中,于4℃冷藏保存,有效期为一个月。该衍生物标准溶液性质稳定,每次使用时,可以直接稀释至所需质量浓度。
2 结果与讨论
2.1 色谱行为
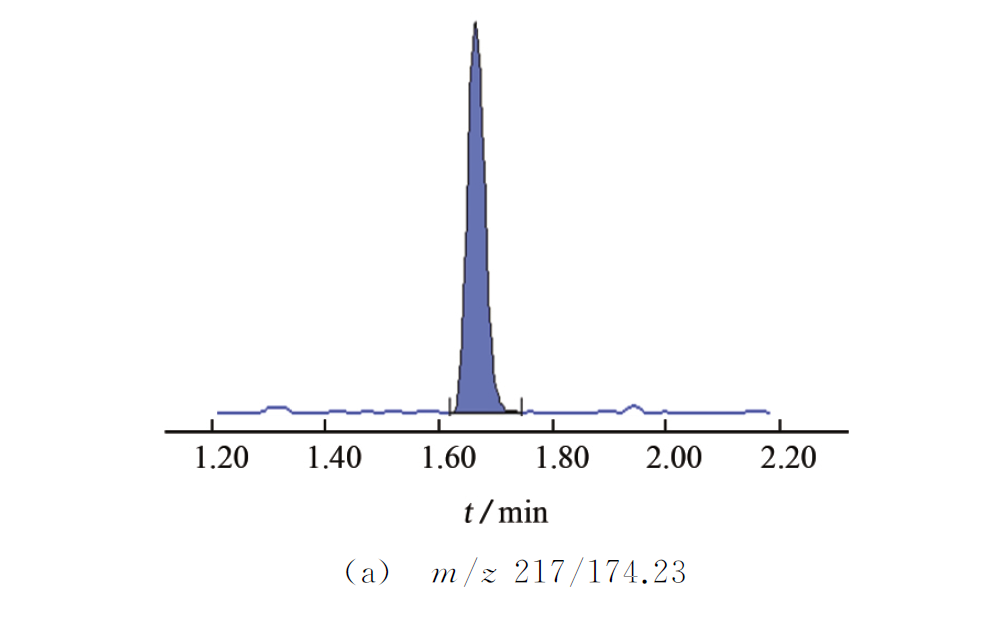
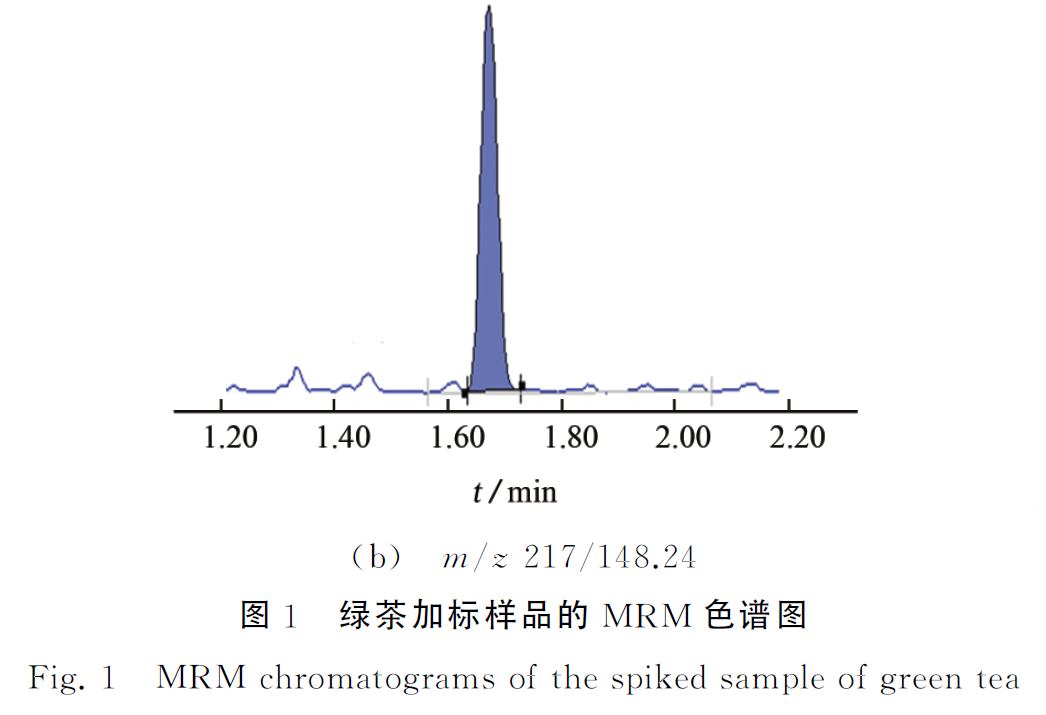
绿茶样品中添加10μg·kg-1 百草枯标准溶液,其MRM 色谱图见图1。
2.2 色谱条件的选择
2.2.1 流动相
试验分别考察了甲醇体系和乙腈体系作为流动相时对测定的影响。结果表明:这两种体系作为流动相时,仪器的灵敏度在同一水平上。考虑到使用甲醇体系系统压力高,试验选择乙腈体系作为流动相。
2.2.2 色谱柱
百草枯极性很强,采用Water BEH UPLC C18色谱柱无法得到保留,一般采用Hillic色谱柱进行分离,为了获得良好的峰形,流动相中必须添加高含量的盐,但高含量的盐容易造成仪器污染。试验将目标物进行了衍生反应,其衍生反应产物在Waters BEH UPLC C18色谱柱上获得了很好的分离效果。试验选用Waters BEH UPLC C18色谱柱。
2.3 质谱条件的选择
2.3.1 母离子
百草枯在碱性条件下被铁氰化钾氧化成1,1-二甲基-[4,4′-联吡啶]-2,2′-二酮,百草枯的衍生反应式见图2。
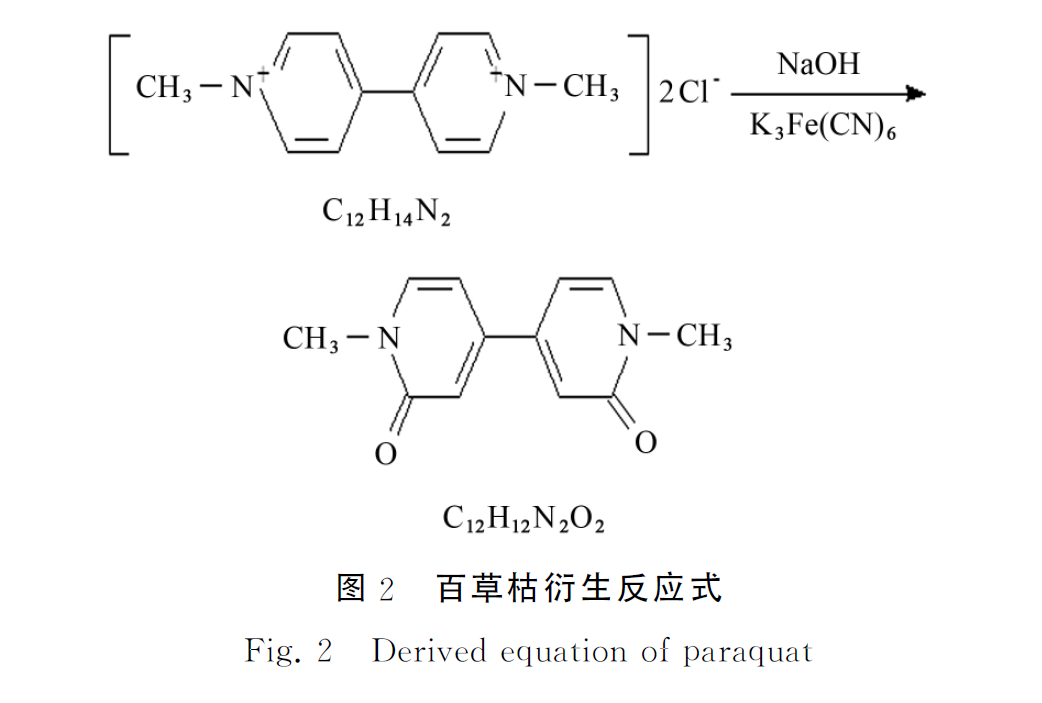
根据百草枯的结构,推断其母离子碎片m/z 应该是217,此推断也在质谱分析中得到了很好的验证。试验选择百草枯衍生物的母离子m/z 为217。
2.3.2 子离子
因衍生反应得到的新物质所在的基质环境复杂,为获得好的质谱图,试验需要先通过色谱柱将新物质与杂质分离,然后再对其进行质谱方法的优化。
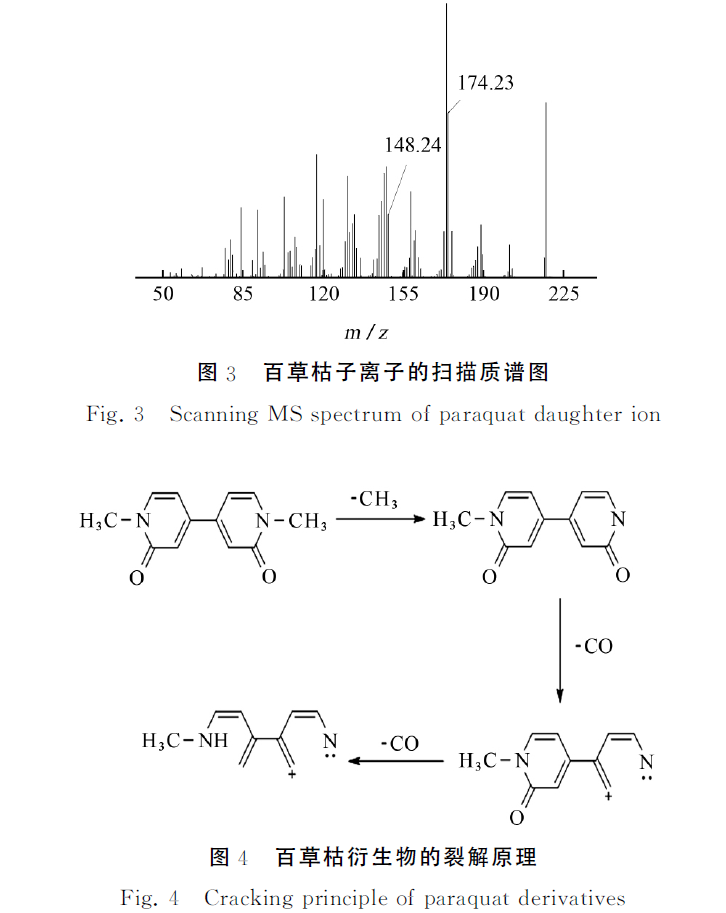
百草枯子离子的扫描质谱图见图3。
由图3可知:174,160,158,148,130,117都是碎片离子,其裂解原理见图4。再通过精确优化确定各子离子的最佳质谱条件,以便获得最佳响应和精确质量数,试验选择m/z 174.23作为定量离子,m/z 148.24作为定性离子。
2.4 标准曲线、检出限和测定下限
按试验方法对百草枯衍生物标准溶液系列进行测定,并绘制标准曲线。结果表明:百草枯的质量浓度在0.001~0.1mg·L-1内与其对应的峰面积呈线性关系,线性回归方程为y=2.100×106x-3.242×102,相关系数为0.9998。
以3倍信噪比计算百草枯的检出限(3S/N)为0.5μg·L-1,以10倍信噪比计算百草枯的测定下限(10S/N)为0.01mg·kg-1。
2.5 方法的重复性和准确性
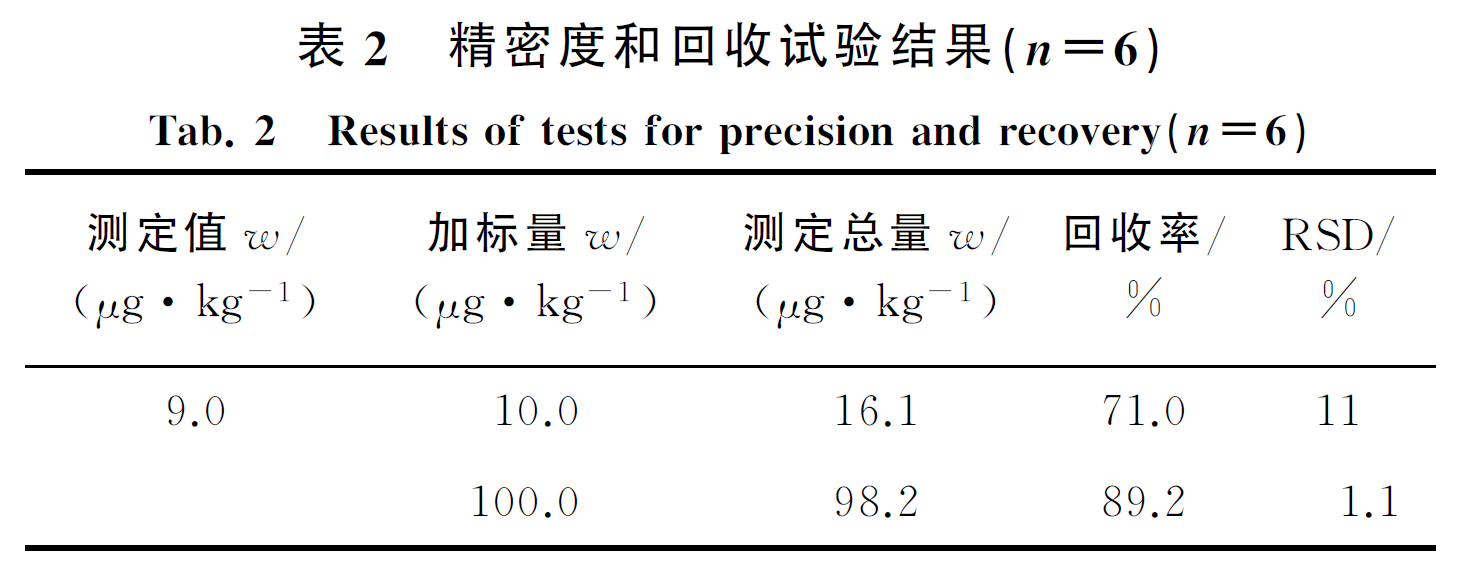
按试验方法对绿茶样品进行分析,并进行加标回收试验,计算测定总量的相对标准偏差(RSD)和加标回收率,结果见表2。
2.6 实验室间数据比较
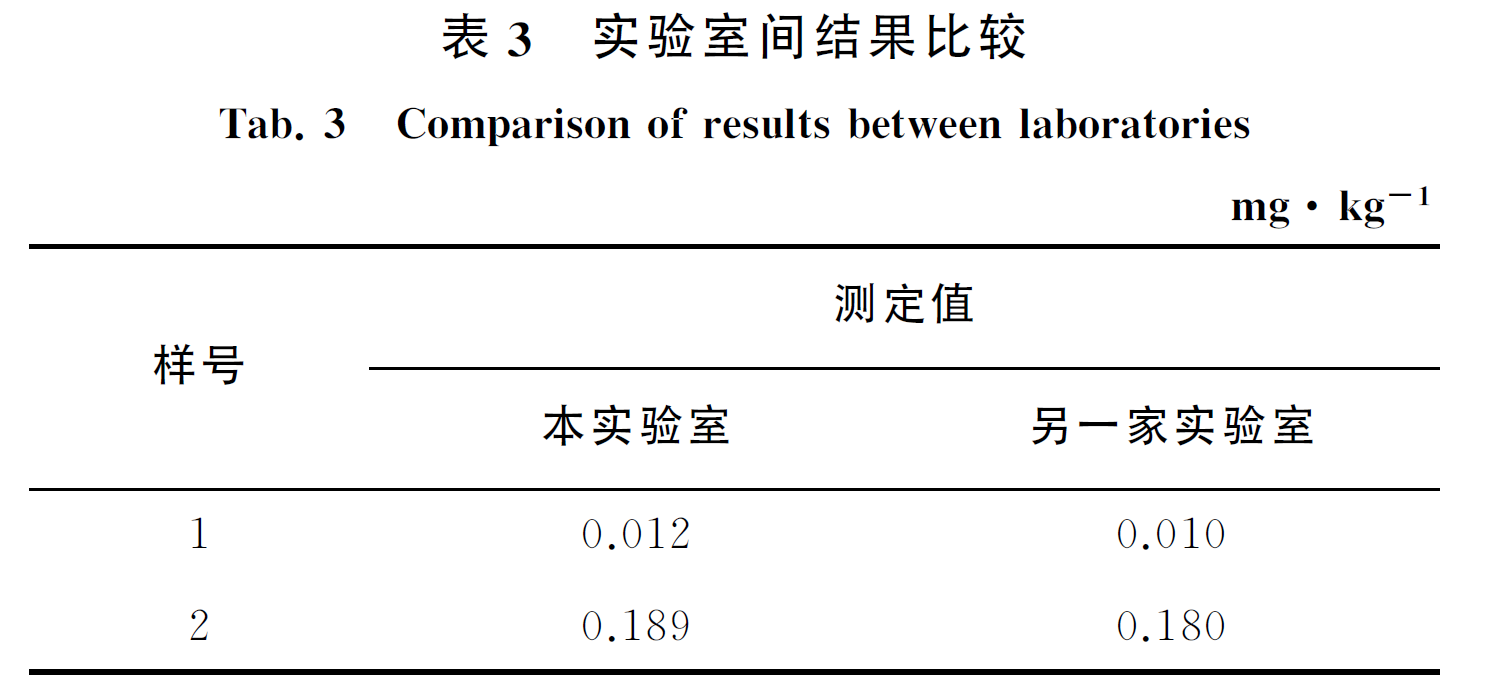
为验证方法的准确性和可靠性,试验将已经粉碎好的700g绿茶样品作为样品1,并制备了一个百草枯理论值为0.2mg·kg-1的阳性绿茶样品(样品2)。
用样品1、样品2进行了实验室间的比对,其结果见表3。
样品若不经过浸泡和磺化处理,直接用水提取后分析,结果是阴性,再次证明了本方法的可靠性,说明百草枯易钝化,直接用水提取很困难。
2.7 样品分析
按试验方法对84个茶叶样品进行分析,其中乌龙茶39个,茉莉花茶16个,绿茶6个,红茶6个,红茶提取物4个,黑茶6个,茶提取物4个,茶浓缩汁3个。结果表明:百草枯测定值高于0.01mg·kg-1的样品有29个,占总样品数的34.5%,百草枯测定值最高为0.22mg·kg-1,百草枯测定值为0.01~0.05mg·kg-1的样品占总阳性结果的82.7%。
本工作采用超高效液相色谱-串联质谱法测定茶叶中百草枯的含量。本方法具有提取效率充分、准确度好、灵敏度高等特点,可满足茶叶类复杂基质的检测要求。
参考文献
[1] 蔡春平,翁若荣,梁鸣,等.AOCO测定农药百草枯含量方法的不足和改进[J].检验检疫科学,2004,14(3):15-17.
[2] CHEN J G,ELDRIDGE D L,LODESERTO F J,etal.Paraquat ingestion:A challenging diagnosis[J].Pediatrics, 2010,125(6):505-509.
[3] ALMEIDA R M D,YONAMINE M.Gas chromatographic-mass spectrometric method for the determination of the herbicides paraquat and diquat in plasma and urine samples[J].Journal of Chromatography B, 2007,853(7):260-264.
[4] 李俊,王震,郭晓关,等.测定鱼腥草中的百草枯和乙草胺残留的三重四级杆气相色谱质谱法[J].山地农业生物学报,2012,31(4):341-344.
[5] 朱震海,宣栋梁.固相萃取-HPLC法测定水中敌草快、百草枯[J].中国卫生检验杂志,2011,21(9):2154-2156.
[6] 季兴繁,邱相君,王勇,等.HPLC法检测人血浆中百草枯的浓度[J].中国药师,2012,15(2):209-211.
[7] 李强,刘海英,王玉红,等.百草枯中毒患者免疫功能改变及临床意义[J].新乡医学院学报,2006,23(6):575-576.
[8] 薄海波.液相色谱-串联质谱法快速测定植物源性食品中的百草枯[J].色谱,2011,29(2):180-183.
[9] 刘萍,邬春华,郑力行,等.生物样品中百草枯检测方法研究进展[J].环境与职业医学,2010(9):563-565.
[10] ZOU T T,HE P L,CAO J J,et al. Determinationof paraquat in vegetables using HPLC-MS-MS[J]. Journal of Chromatographic Science,2015,53(2):204-209.
[11] PIZZUTTI I R,VELA G M E,KOK A D,et al.Determination of paraquat and diquat:LC-MS method optimization and validation[J].Food Chemistry, 2016,209(15):248-255.
付萌、赵春华、张伟伟、邓锁成
梅里埃营养科学(中国) 诺安实力可
